FTMove Help
Welcome to FTMove! This page offers support for each facet of the FTMove server, from job submission to analysis of results. Use the links below to jump to your topic of interest. Please feel free to contact us if you have any issues.
-
Login
Job Submission - PDB Input
Job Submission - Upload Structures Input
Domain Splitting
Queue
Job Results
Login Page
When you arrive at the FTMap page, the first screen you see is the login screen, shown here:
- If you already have a username and password, fill in the boxes and click Sign in. Proceed to Job Submission.
- If you prefer not to use an account, select "Use the server without the benefits of your own account". Know that your job submissions will be publicly accessible.
- If you would like to sign up for an account, click the Sign Up option in the top right corner of the screen. Proceed to Sign Up.
- If you are a returning user, but have forgotten your password, click Forgot Password? to have a new password emailed to you
Job Submission - PDB Input
This is the job submission page for FTMove, with the 'Job Input Type' button toggled toward PDB input. See below for details on job submission with PDB input.
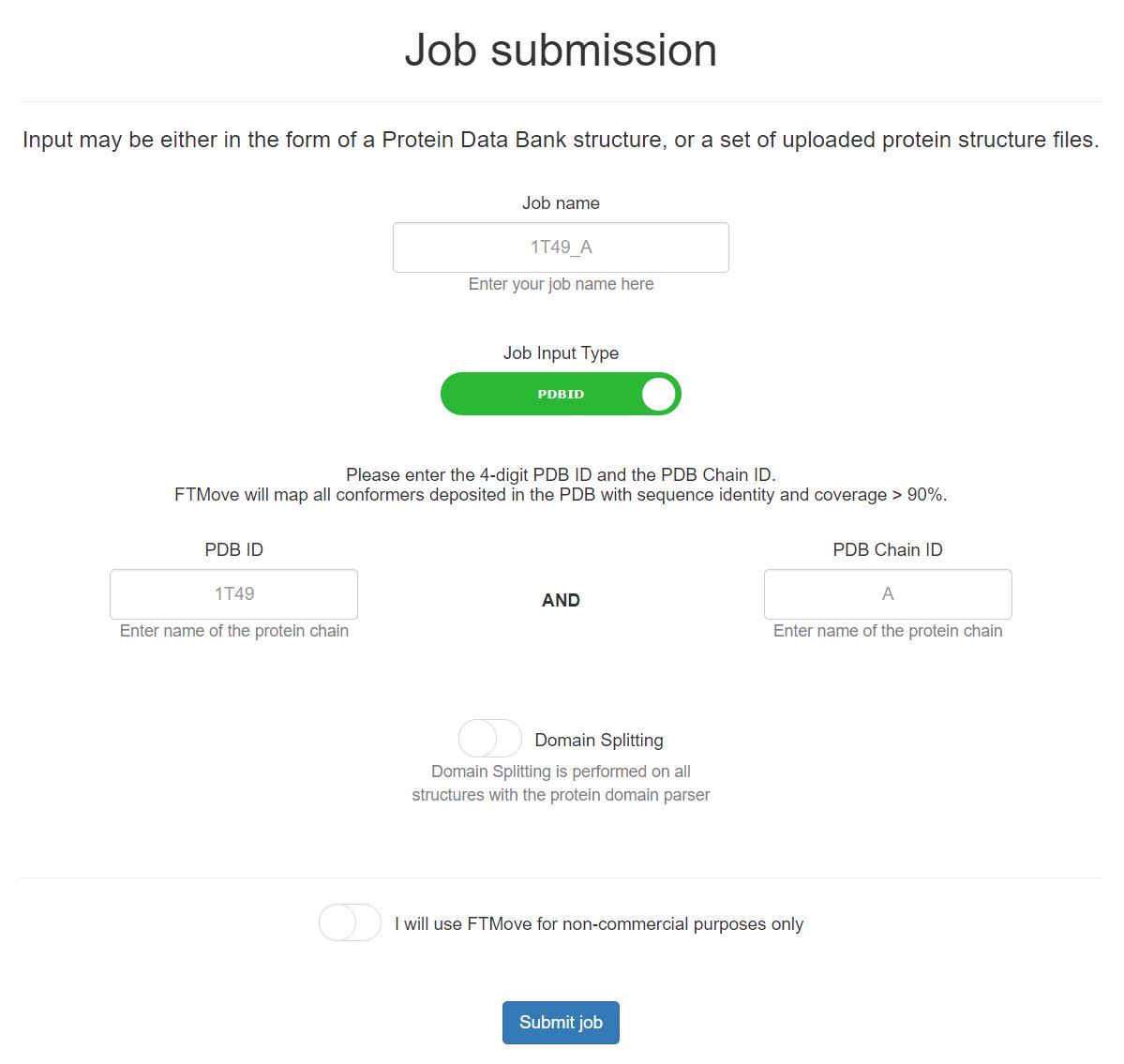
- Job Name: Provide a job name for this submission. If you choose to leave this blank, a unique ID will be created for this field.
- PDB ID: Enter the 4-digit PDB ID, uppercase or lowercase.
- PDB Chain ID: Enter the PDB Chain ID, in the same case as listed by the PDB. At this time, FTMove does not support mapping of two or more chains at once.
- Domain Splitting: The domain splitting button default it set to OFF. Please reference the Domain Splitting section to decide whether this feature should be utilized in your job submission. To select the domain split feature, click the button and the background will turn green.
- How it works: FTMove will identify all structures in the PDB with sequence similarity > 90% and sequence coverage > 90%, via searching with MMSeqs2. Each indivudal structure will be mapped with the algorithm described by FTMap, which removes all ligands, non-standard amino acid residues, and small molecules prior to mapping.
Job Submission - Upload Input
This is the job submission page for FTMove, with the 'Job Input Type' button toggled toward upload input. See below for details on job submission with structure uplaoded as the input.
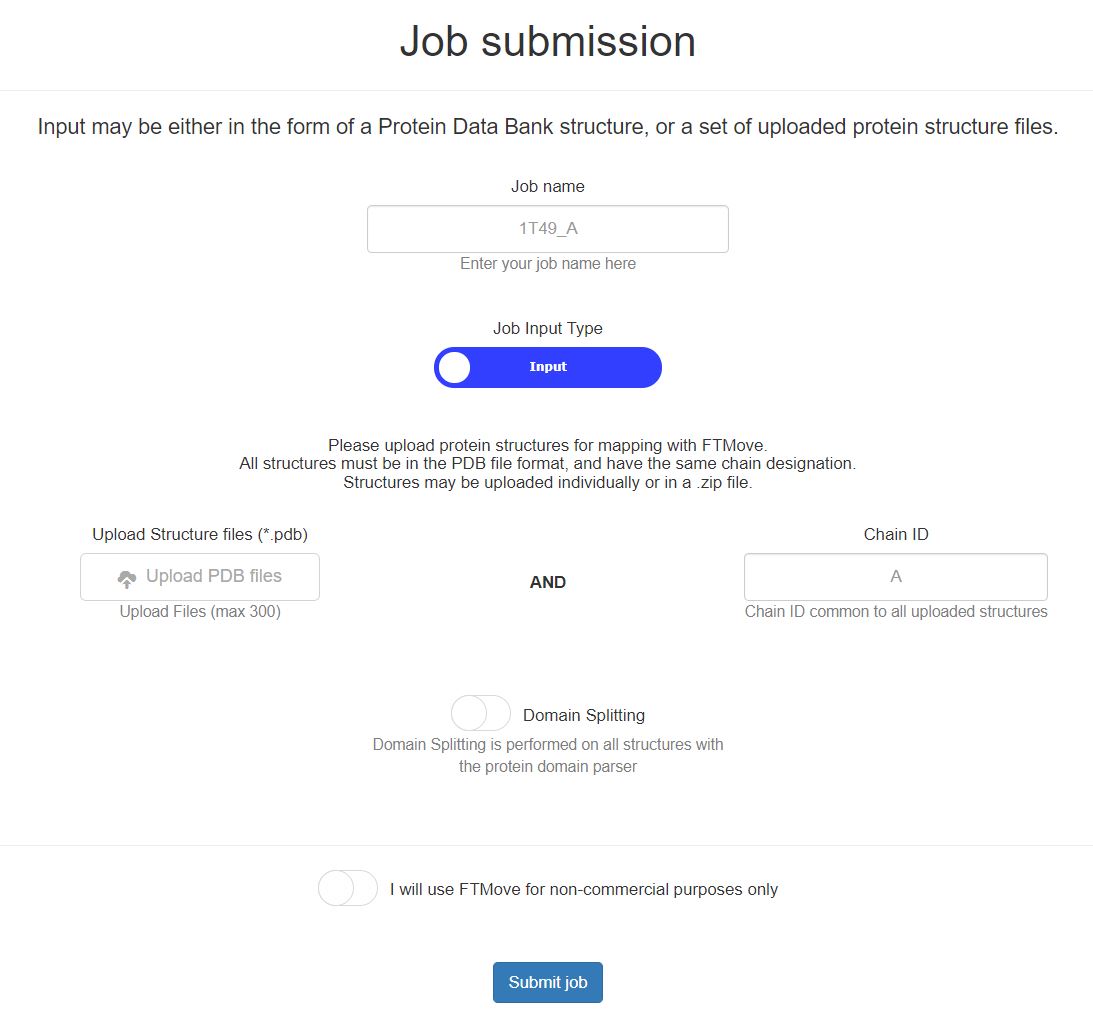
- Job Name: Provide a job name for this submission. If you choose to leave this blank, a unique ID will be created for this field.
- Structure Upload: Upload the protein structure PDB files, each file must be in the PDB format and have a .pdb extension. Files can be uploaded individually or a zip file of pdb files may be uploaded. Each structure must have a unique file name.
- Chain ID: Enter the Chain ID common to all structures uploaded. At this time, FTMove does not support mapping of two or more chains at once.
- Domain Splitting: The domain splitting button default it set to OFF. Please reference the Domain Splitting section to decide whether this feature should be utilized in your job submission. To select the domain split feature, click the button and the background will turn green.
- How it works: FTMove will map each uploaded structure with the algorithm described by FTMap, which removes all ligands, non-standard amino acid residues, and small molecules prior to mapping. For easiest analysis of results, it is best to keep the structure file names short, with the unique identifier at the begining of the file name.
Domain Splitting
The domain split option is included for your convenience. We reccomend using the domain splitting feature when the protein is fairly large and made up of two or more distinct domains. Domain splitting is performed by the Protein Domain Parser (PDP) on each protein structure mapped in the job.
One advantage of domain splitting prior to mapping is that that probes will be more concentrated on each domain. For example, consider a typical kinase with two domains. When the both domains are mapped together, most probes have low binding energy to residues surrounding the ATP binding pocket. However, when each domain is mapped individually, there are twice as many probes distributed across the entire protein, with half on each domain. As a result, sites that are far from the ATP binding site typically are typically observed 'stronger' (i.e. having more probes bound), relative to mapping the complete structure. Thus, domain splitting permits more accurate mapping of the non-ATP binding region.
One caution with domain splitting is that by mapping each domain seperately, FTMove may identify binding sites between the domains that may not exist biologically. Therefore, interpret any inter-domain binding sites with caution, and compare mapping results between domain split and non-domain split structures to determine if such sites are biologically relevant.
- If all structures were split into simlar domains, a green message will appear in the results page once your job has completed, indicating all structures were split into X number of domains. (If all structures have only 1 domain, the results are the same as if domain splitting was not applied).
- If domain splitting resulted in a different number of domains across all structures, a red message will appear in the results page once your job has completed. The message will indicate how many structures were split into domains, relative to the total number of structures. Additionally, the number of domains in each structure will be included in the output table displayed with the job results. To ensure all domains are split equally, you may perform the domain splitting yourself, and use the Job Submission - Structure Upload feature to assess each domain, one at a time.
Queue Page
Use this page to see the status of your submitted jobs. Jobs are run in order they were submitted. Runtime will vary by the job. Each status is explained in the table below
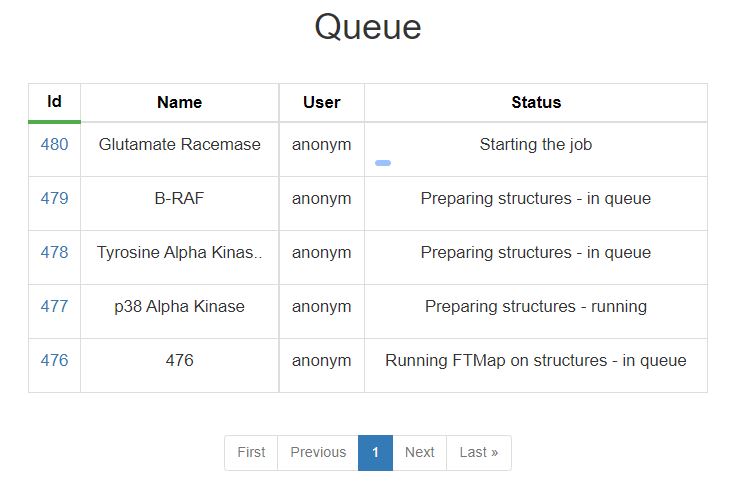
| Status | Description |
|---|---|
| Starting the Job | Processing job input and preparing for submission, this step is very fast |
| Preparing Structures | This step searches for similar protein structures (if applicable), aligns all structures, and prepares setup for mapping. This step is either in queue or running, and is typically completed fairly quickly |
| Running FTMap on Structures | All individual structures are mapped with the algorithm described by FTMap. Depending on the number of structures in the job and server availability, this step may take up to a few days. |
| Evaluating Binding Sites | The mapping resluts of all structers are used to determine and rank the binidng sites. |
| Job Completed | Your job has finished successfully, click on the job number to view the results. |
| Error | An error has occured during the processing of your job, please contact us if you need help troubleshooting. |
Results Page
Use this page to view the results of your completed job. The top 10 ranked binding sites are shown in the web page with NGL viewer. Additionally, the structure-binsing site strengths are reported in a downloadable table.
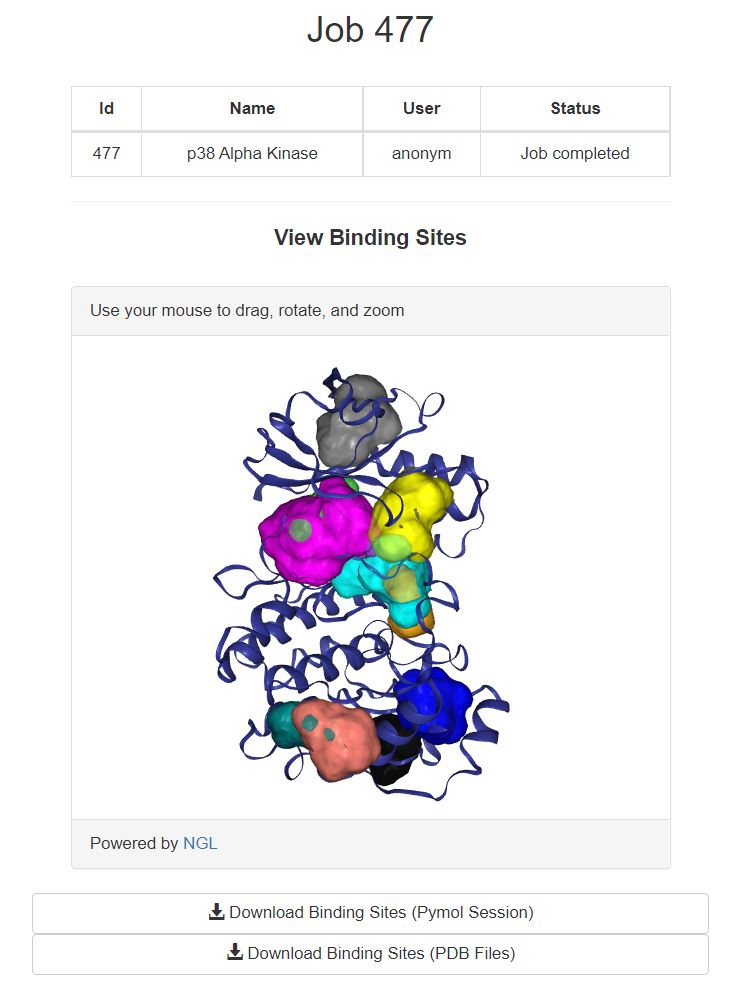
The top 10 binding sites shown here are colored according to rank. Starting with the highest ranked site (000 - cyan) > (001 - magenta) > (002 - yellow) > (003 - salmon) > (004 - gray) > (005 - blue) > (006 > orange) > (007 - limegreen) > (008 - teal) > (009 - black). If more than ten binding sites were identified, they can be viewed by downloading the binding sites.
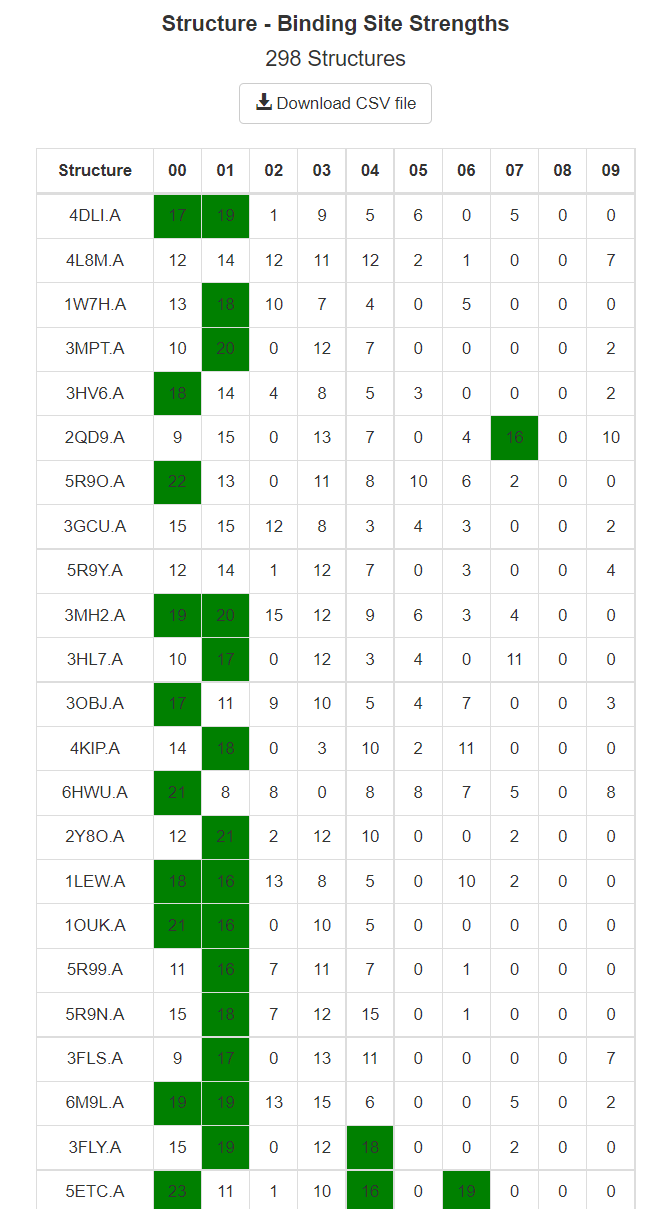
This table shows the Structure-Binding Site Strenths. For each individual structure mapped, the number of probe clusters within 2 Angstroms of the binding sites are tabluated and shown here. For example, in the above image mapping of the PDB structure 4DLI.A found 17 probe clusters in the top binding site (000), and 19 probe clusters in the second strongest binding site (001). Again, strengths for only the top 10 binding sites are resported in the web-page, and the strengths for all binding sites are available in the CSV, accesablie by the 'Download CSV file' link shown at the top of the table.
Additional download options are available at the bottom of the results page.
- Download Structure FTMap Results (pymol session): This pymol session the FTMove-identified binding sites, and each structure mapped, along with their mapping-identified probe clusters. The structures are aligned to the input PDB ID or to the first of many structures uploaded, using the pymol align function. Visual representation of the number of probe clusters reported in the table (decribed above), as the actual structure-binding site strengths are based on a local alignement of the mapping consensus sites. This file can get quite large, and may take a while to open on your computer. Pymol version 1.8.5.0 or greater is required to load all feature of the pymol session.
- Download Structure FTmap Results (tar.gz. of PDB files): This download (ftmap_files.tar.gz) contains all of the individual structure mapping results. If homologs were obtained from the PDB, the files are named as PDB_ID.CHAIN_aligned_ftmap.pdb and if structures were uploaded to FTMove they are names FILE_NAME_aligned_ftmap.pdb. The ftmap PDB files have separate headers for the 'protein' mapped, and each consensus site (crosscluster), in order for increasing strength.
- Download Clustered Heatmap (PNG File): This clustered heatmap was generated with the correlation distance metric, using the structure-binding site strengths for all binding sites. Clusters formed with the 'average' joining method. The raw data used to create this table is available by downloading the csv of the structure-binding site table.
Vajda Lab (Boston University).